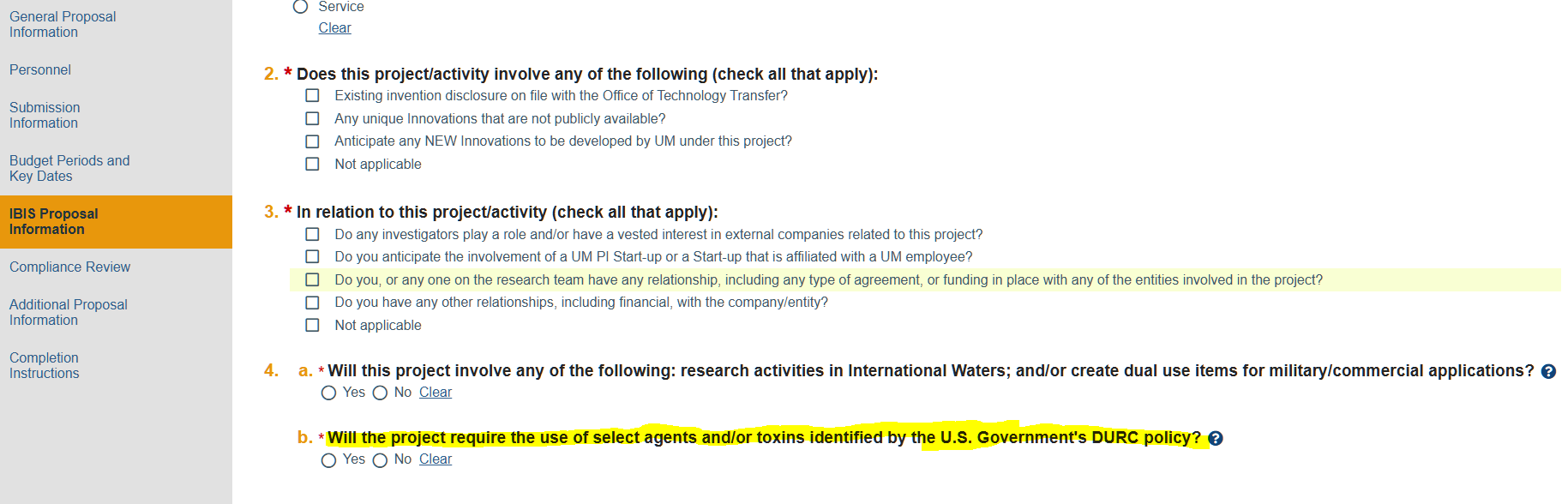
System Upgrade & Downtime:
UDisclose System and IBISResearch Suite – IRB, Grants, Agreements, IACUC, and AOPS
Dear Research Community,
The Office of the Vice Provost for Research & Scholarship (OVPRS) is excited to announce the upgrade of the UDisclose System and IBISResearch Suite to v10.5!
For more information about the new features that will be part of this upgrade, please visit the Huron 10.5 Upgrade website. This website serves as a resource repository for users impacted by this upgrade. You will be able to readily access release notes, contact information, key project milestones, and software version transition videos.
Please take note of the details below:
Start Date: Friday, June 27th, 2025, at 6:00 PM
End Date: Monday, June 30th, 2025 at 8:00 AM
Downtime is estimated to be about 60 hours, and will affect other areas of research, including the Velos D-link, dashboards, and other reporting systems that communicate with IBISResearch.
This interruption is a necessary step to ensure a smooth transition to our new and improved research system. We appreciate your understanding and cooperation as we work towards enhancing our research infrastructure.
Cleanup Effort
What to Do
As we prepare to upgrade our IBISResearch system this summer, we are undergoing cleanup efforts in the current system to align with more accurate reporting and data integrity for future state.
Some of these may be done administratively by our office (cosmetic changes, etc.). However, to ensure that the upgraded system has all of the information we will need, we need your help to ensure that your studies reflect the most up-to-date information, including an accurate list of study team members.
This includes removing personnel that are no longer actively working on the study and/or have an inactive IBISResearch account. If corrections are needed, please submit a modification as soon as possible. If an account needs to be reactivated, please contact OVPRS Help Desk at ovprshelpdesk@miami.edu or call 305-243-2314.
In addition, this is a good opportunity to review some of the custom reports that exist in each solution (by navigating to the Reports sub menu, and clicking on the Custom Reports tab). If there are any reports that need to be updated, please notify us via our Support Portal.
How Can You Prepare?
Plan Accordingly
In preparation for the upgrade, we will continue to provide features and action items in the coming weeks.
In the meantime, we recommend saving/completing/submitting any pending work or submissions in the UDisclose and IBISResearch systems before the downtime begins on June 27th at 6:00 PM.
For IRB: Please contact the assigned IRB coordinator if there is any urgency associated with the review of your submission(s) in order to have any outstanding issue(s) resolved, if possible, prior to the IBISResearch downtime.
The IBISResearch systems will be inaccessible during the timeframe mentioned above, and there will be no access to IRB-approved study documents in the Velos D-Link. If there are any important study updates needed prior to this downtime, please be sure to submit these in IBISResearch system as soon as possible. Otherwise, updates will resume once the IBISResearch system is live on Monday, June 30th, 2025.
If there is any urgent item (impacting subject safety) that needs IRB consideration during the downtime, please contact an HSRO manager (dding@miami.edu) via email with a detailed description. You will be contacted for any follow-up.
Submissions that are received closer to the downtime are likely to experience a delay in review time.
For Grants: For anything due on June 27th, please reach out to Deborah Musgrove (dmusgrove@miami.edu)
For Agreements: For anything due on June 27th, please reach out to Holly Kasem-Beg (holly.kasem-beg@miami.edu).
For COI: Submissions of disclosures and Pre-Approval Requests (PARs) will not be possible during this downtime window. If your Disclosure Profile is currently in Action Required state, please submit it as soon as possible.
Disclosures that are completed closer to the downtime are likely to experience a delay in review time.
For IACUC: For anything due on June 27th, please reach out to Lizzeth Meza (lmeza@med.miami.edu).
For AOPS: For anything due on June 27th, please reach out to Dale Wincenciak (dwincenciak@med.miami.edu)
If you have any questions about the upgrade, please contact:
Go-Live Support
As we gear up for this exciting upgrade on June 30th, please remember that our support team will be available to assist you with any questions or issues you may encounter.
Human Subjects Research Office (305-243-3195; hsro@miami.edu) is available to answer any questions regarding the IRB process during the downtime.
UDisclose System Helpline (305-243-0877; dsam@miami.edu) is available to answer any questions regarding the disclosure process during the downtime.
Compliance Inbox (305-243-6296; compliancehelp@miami.edu) is available to answer any questions regarding the Pre-Approval Request process during the downtime.
IACUC: Please contact IACUCsupport@med.miami.edu if there is any urgency associated with the review of your submission in order to have the issue resolved, if possible, prior to the IBISResearch temporary downtime.
AOPS: Please contact dvr@med.miami.edu if there is any urgency associated with the review of your submission in order to have the issue resolved, if possible, prior to the IBISResearch temporary downtime.
Need Help?
Contact Us.
For any system issues, questions, or general assistance, contact the OVPRS Help Desk