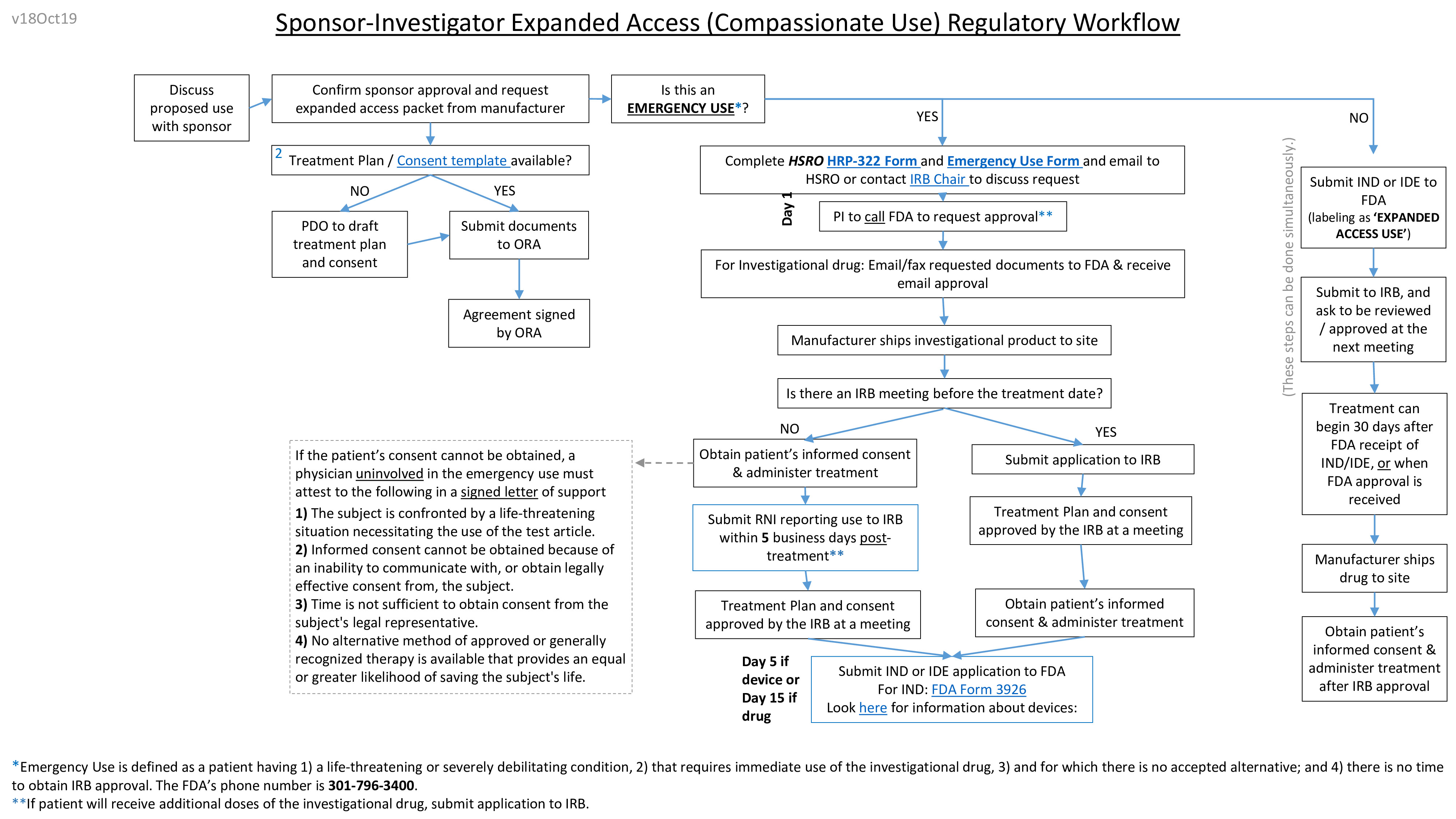
“Emergency Use” is a provision of the FDA regulations that allows a physician to use an investigational product in a life-threatening emergency when there isn’t sufficient time to obtain IRB review and approval.
When seeking to use an unapproved product, it is critical that the patient and his/her licensed physician consider all possible risks because the FDA has not determined whether the products are safe. Such products may, or may not, be effective in the treatment of the condition. See more details at: https://www.fda.gov/news-events/expanded-access/expanded-access-information-physicians
Check to see if the regulatory criteria for use of the investigational product without IRB approval are met by reviewing Worksheet Emergency Use (HRP-322).
When its decided that the investigational product is the best option for the patient in an emergency, physicians must complete the following steps.
- Contact the sponsor and obtain authorization
- Contact the FDA to obtain approval.
- When time permits, submit the following to IRB via email to an HSRO analyst or directly to an IRB chair to discuss the use.
- Patient’s current disease or condition - including the justification of the use
- Treatment plan/protocol
- Sponsor’s authorization
- Consent template (HRP-502) -hrp-502h---template-consent-document--expanded_access.docx
- eIND (if the use involves investigational drug)- FDA email approval
- IB or device manual/information